MALVIRUS
MALVIRUS is a fast and accurate tool for genotyping haploid individuals that does not require to assemble the read nor mapping them to a reference genome. It is tailored to work with virological data (including but not limited to SARS-CoV-2) and can genotype an individual directly from sequencing data in minutes.
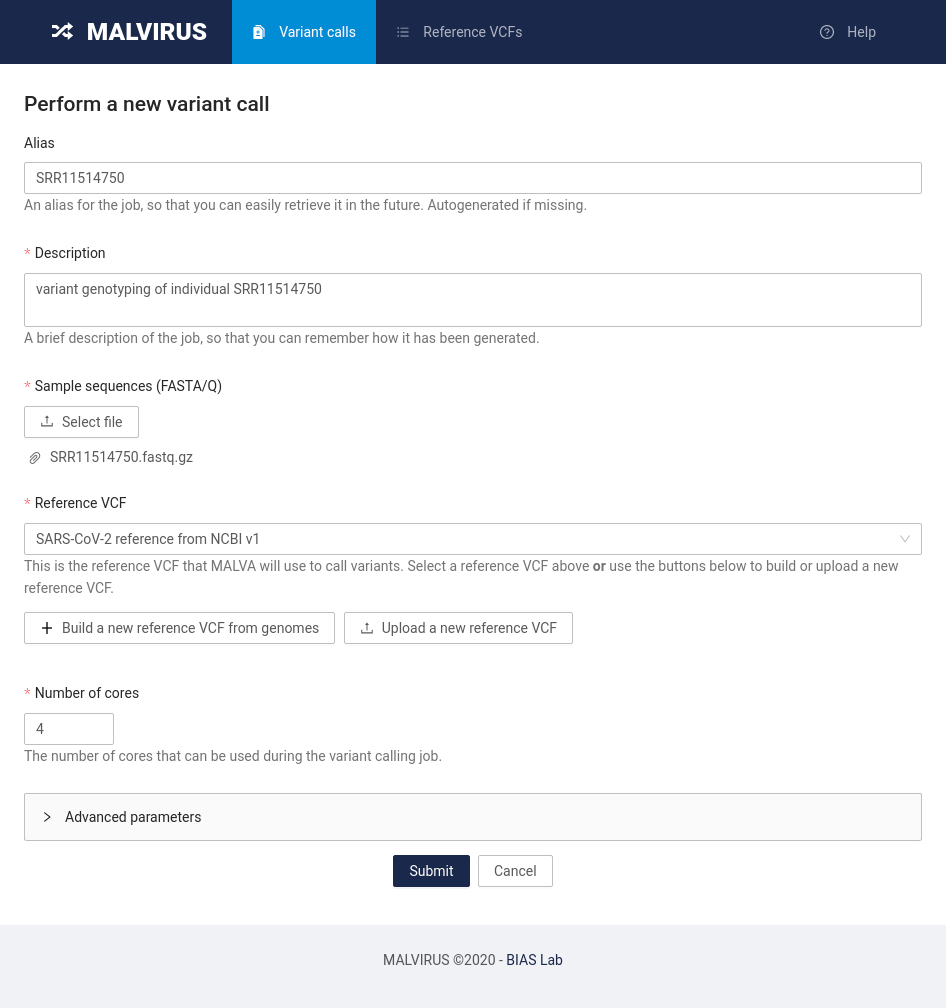
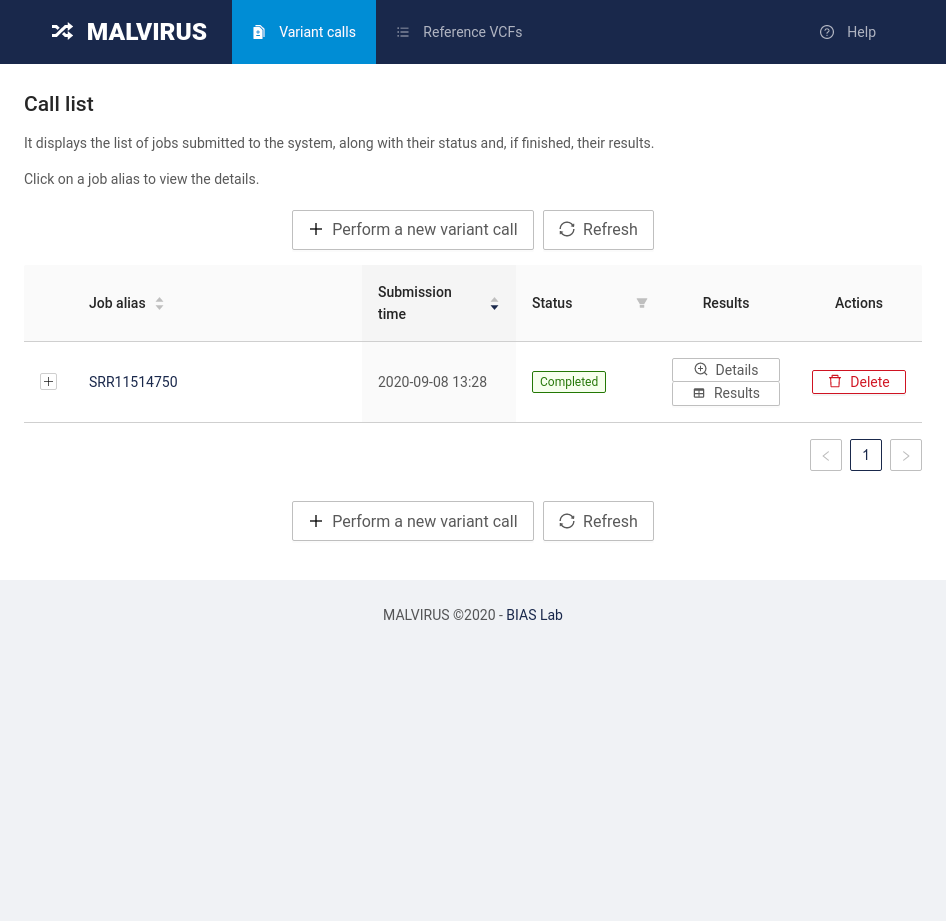
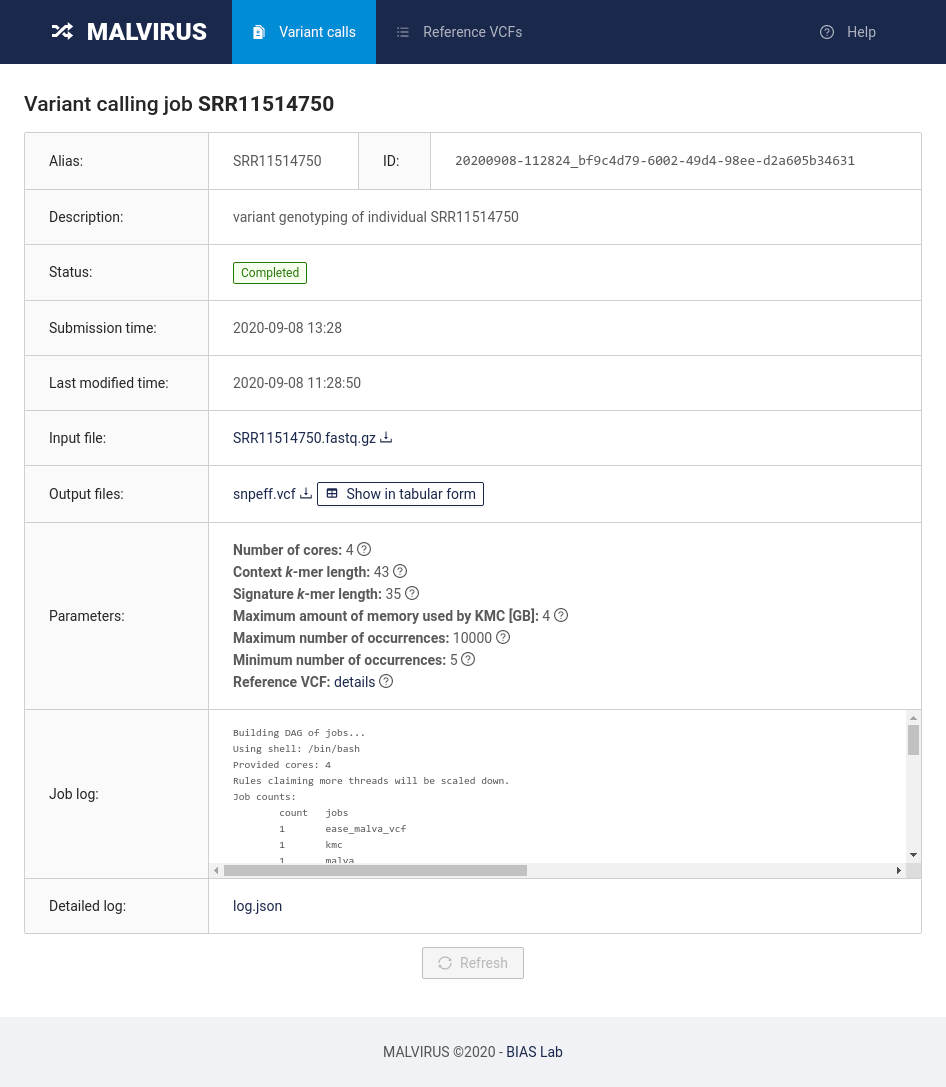
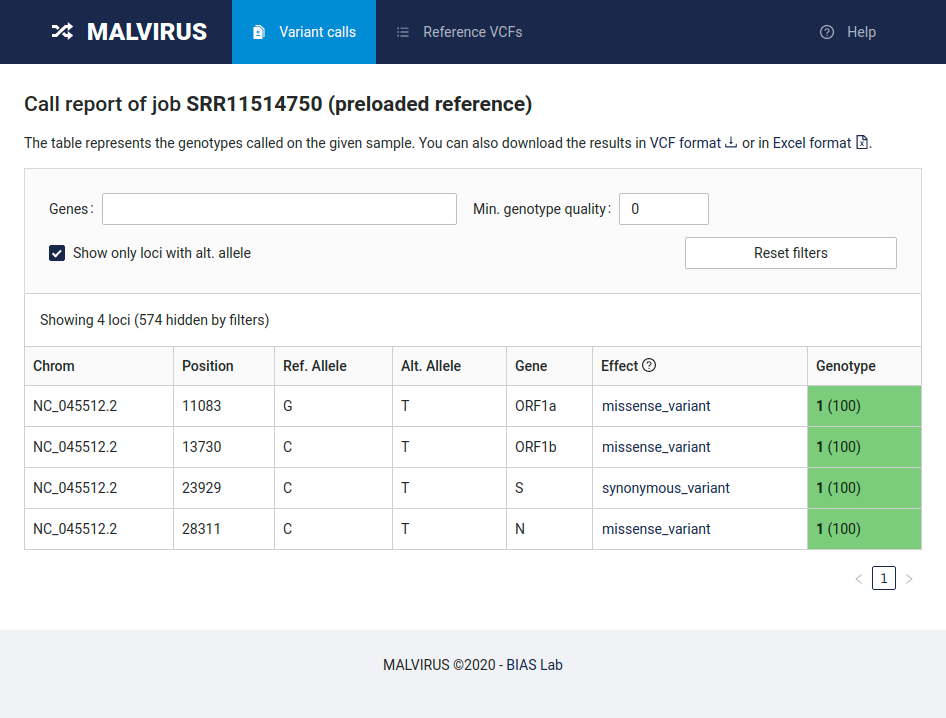
MALVIRUS is divided into two logically distinct steps: the creation of a variant catalog from a set of assemblies and the genotype calling. The first step is based on mafft [1] and snp-sites [2], whereas the second step is based on KMC [3], MALVA [4], and SnpEff [5].
The variant catalog can be built once and reused for genotyping multiple individuals.
Please see the following documents for additional details:
MALVIRUS is distributed as a Docker image and is publicly available on GitHub and Docker Hub under the terms of the GNU General Public License version 3 or later. MALVIRUS was mainly developed and tested under Ubuntu GNU/Linux version 18.04 but works wherever Docker is available.
Citation
MALVIRUS: an integrated web application for viral variant calling
Simone Ciccolella, Luca Denti, Paola Bonizzoni, Gianluca Della Vedova, Yuri Pirola, Marco Previtali
bioRxiv 2020.05.05.076992; doi: 10.1101/2020.05.05.076992
References
[1] Katoh, Kazutaka, and Daron M. Standley. 2013. “MAFFT Multiple Sequence Alignment Software Version 7: Improvements in Performance and Usability.” Molecular Biology and Evolution 30 (4): 772–80. doi:10.1093/molbev/mst010.
[2] Page, Andrew J., Ben Taylor, Aidan J. Delaney, Jorge Soares, Torsten Seemann, Jacqueline A. Keane, and Simon R. Harris. 2016. “SNP-Sites: Rapid Efficient Extraction of Snps from Multi-Fasta Alignments.” Microbial Genomics 2 (4). doi:10.1099/mgen.0.000056.
[3] Kokot, Marek, Maciej Dlugosz, and Sebastian Deorowicz. 2017. “KMC 3: counting and manipulating k-mer statistics.” Bioinformatics 33 (17): 2759–61. doi:10.1093/bioinformatics/btx304.
[4] Denti, Luca, Marco Previtali, Giulia Bernardini, Alexander Schönhuth, and Paola Bonizzoni. 2019. “MALVA: Genotyping by Mapping-Free Allele Detection of Known Variants.” iScience 18: 20–27. doi:10.1016/j.isci.2019.07.011.
[5] Pablo Cingolani et al. 2012. “A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3” Fly 6(2): 80-92. doi:10.4161/fly.19695.